문과 아재도 쉽게하는 R 데이터 분석 – (2) kNN (k nearest neighbor)
k Nearest Neighbor
고정관념과 범주화
제일 가까운 이웃이라는것은 무엇일까요? 가까운 이웃들을 보고 그것도 그 범주에 넣어버린다는 뜻입니다. 즉, 유유상종이라고 그사람을 보려면 어울리는 친구를 보라라는 말마따나, 친구들이 소위 날라리면 그사람도 날라리겠거니 판단하는 겁니다.
사람들이 겪는 경험적 현상을 그대로 생각하면 되는데,
예를들어 인간은 정말 적은 표본에도 기존 경험으로 빠르게 무언가를 판단하는 경향이 있습니다.
그동안 동물이 진화해오면서 DNA에 적혀있는 부분 (표정 등), 그리고 후천적으로 습득한 자기 경험에 의해 (특정 나라에 대한 편견 등), 몇개의 표본만을 보고 저사람은 XX 일것이라는 판단을 하게 됩니다.
머신러닝에도 이렇게 판단하는 기법이 있습니다.
kNN(k Nearest Neighbor)가 바로 그렇습니다.
산점도로 살펴보기
예를들어 아래와 같은 산점도를 한번 생각해보세요.
데이터는 머리카락의 길이와 키의 크기를 모았고, 이에대해 성별을 구별하고 있습니다.
이러한 상황에서 특정 머리카락의 길이와 키의 크기를 알고 있는 상태에서 점을 한번 찍어 봅니다.
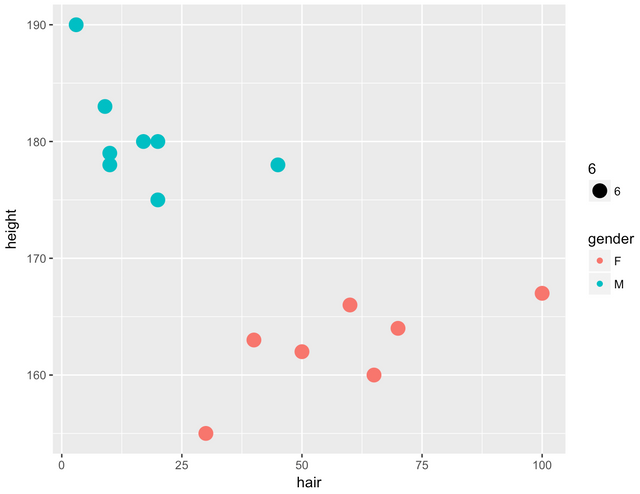
그런다음 그 점 주위의 다른 점들이 남자인지 여자인지 확인해봅니다. 확실히 머리카락 길이처럼 성별간 유의한 경향성이 있다면 정말 단순한 생각이지만 머리카락이 길면 여자구나, 라고 판단하는게 빠르고 간편한 방법일수 있을것입니다. kNN은 그냥 주변의 점을 참고로 해서 결정하는 정말 간단한 방법입니다.
k는 몇개까지 확인해보는지에 대한 수인데, k가 1이라면 제일 가까운 점으로 우리의 목표값도 그럴것이구나 결론을 내어버리게 됩니다. k가 10이라면 주위의 제일 가까운 점 10개를 보고 제일 많은 범주로 분류해버리면 됩니다.
만약 키가 190이고, 머리카락의 길이가 5cm인 스포츠머리를 남자로 판단해야할지 여자로 판단해야할지 모른다면 일단 이러한 수치를 가지고 있는 데이터들의 성별을 살펴보는 것이죠. k가 10이면 주위 10개를 보겠고, 가까운 점들은 (키:188, 머리카락:10cm, 남자) , (키:192, 머리카락:2cm, 남자) 와 같은 데이터들이겠죠.
물론 이러한 방법은 항상 맞지 않습니다. 현실세계가 그러한것처럼 체격이 크다고 해서 모두 남자도 아니고 머리카락이 길다고 해서 항상 여자도 아닙니다. 다만 기계적으로 그렇게 데이터가 분포되어있기때문에 그렇게 분류가 되는 것입니다.
K가 작으면?
k가 1이라면 정말 미묘한 노이즈에서도 민감하게 반응하게 됩니다. 예를들어 우연히도 데이터가 여자농구선수 근처에 존재한다면, 성별은 남자로 구별되어야함에도 특이한 점을 따라갑니다. 그리고 엉뚱하게 분류될 가능성이 많아집니다.
K가 크면?
k가 극단적으로 크다면, 결국에는 데이터에서 제일 많은 비율을 차지하는 성별로 분류가 됩니다. 남자가 더 많았다면 남자로, 여자가 더 많았다면 여자로 분류가 될것입니다.
그렇다면 k는 어떻게 결정하여야 하나?
K를 하나씩 변경해보고 테스트데이터에서 최적의 성능을 내는 값으로 바꿔주는것이 필요합니다. 보통 3~10사이로 결정하며, 전체 데이터수의 제곱근으로 계산하는 방법도 존재합니다.
실제 테스트 해보기
실제, 몇가지 연습용 샘플이 있습니다. UCI의 기계학습 저장소를 살표보면 유방암에 대한 자료들이 있습니다.
아래의 URL에서 데이터는 확인할 수 있습니다.
- https://archive.ics.uci.edu/ml/machine-learning-databases/breast-cancer-wisconsin/
- https://github.com/stedy/Machine-Learning-with-R-datasets
> wbcd <- read.csv("wisc_bc_data.csv")
> str(wbcd)
'data.frame': 569 obs. of 32 variables:
$ id : int 87139402 8910251 905520 868871 9012568 906539 925291 87880 862989 89827 ...
$ diagnosis : Factor w/ 2 levels "B","M": 1 1 1 1 1 1 1 2 1 1 ...
$ radius_mean : num 12.3 10.6 11 11.3 15.2 ...
$ texture_mean : num 12.4 18.9 16.8 13.4 13.2 ...
..이하생략..
다른것들은 암을 판단하기 위한 여러가지 특성들이 되겠고, 결국 구하고자 하는것은 diagnosis가 되겠습니다. B와 M은 Benign(정상), Malignant(비정상)로 암의 유무가 되겠습니다.
일단, 어떤 데이터가 정상인지 비정상인지 우리에게는 이미 답이 있기 때문에, 새로들어오는 데이터들은 그에대해 제일 가까운곳으로 가이드만 주면 됩니다. 다만 참고할 점의 갯수인 k만 결정하면 되는것이죠.
데이터의 구조
데이터 프레임이기 때문에 바로 훈련에 이용해볼수 있습니다.
이 데이터가 kNN을 이용하기 왜 좋은지 살짝 살펴보면, 우리가 위에서 예를들었던 성별구분문제와 마찬가지로, 판별에 필요한 데이터는 수치형이고 구분해야하는 속성은 명목형이기 때문입니다. 여러가지 수치를 가지면 공간상 어떠한 점에 있는지 수치적으로 계산이 가능하며, 이를통해 가까운 점에 대한 계산이 용이하게 됩니다.
예를들어, 키가 182인 데이터에 가까운 점들을 찾으려면 내림차순을 해서 대충 182와 차이가 적게 나는 놈들부터 뽑아내면 되겠죠. 만약 이 데이터가 범주형이면 바로 사용하기에는 녹록치 않습니다.
암덩어리 데이터도 마찬가지로, 암의 유무는 명목형 자료이며 나머지는 수치적 자료입니다. 이는, 바로 kNN을 적용시켜볼수있다는 뜻이 되겠습니다.
데이터 자르기
데이터는 항상 반 이상은 훈련용으로 쓰더라도 어느정도는 테스트용으로 남겨두어야 합니다. 훈련을 시킨 데이터로 다시 테스트를 한다는것은 의미가 없습니다. 예를들어 과거의 주식가격으로 열심히 훈련시킨 다음에 다시 과거를 예측하면 당연히 거진 다 맞겠지만, 우리가 원하는건 언제나 잘 작동하는 예측모델이기 때문입니다.
따라서 데이터를 다음과 같이 잘라보겠습니다. 569개가 있으니 69개만 테스트 데이터로 쓰겠습니다.
train <- wbcd[1:500,c(-1,-2)]
test <- wbcd[501:569,c(-1,-2)]
1행과 2행을 제외하는 이유는, 훈련에 굳이 필요하지 않은 데이터이기 때문입니다. 훈련에 필요한 열을 제대로 지정해주지 않으면 모든 대상 열이 다 포함되게 되는데 이럴때는 부득이하게 ID역할을 하는 1번 열이라든지, 결과가 들어있는 diagnosis 이름의 열 (2번열)은 제외하고서 훈련시켜야 합니다.
데이터 예측하기
이제, knn을 통해 암덩어리를 예측해볼수 있습니다.
사실 보통의 경우는 모델이 튀어나오고 이 모델을 통해 predict() 함수를 사용하여 예측하고는 합니다. 하지만 knn은 바로 train데이터와 test데이터를 넣어서 예측한다는 점이 다른점인데, 이는 사실 knn이 모델이랄게 없기 때문입니다. 그냥 기존 데이터에 테스트 데이터를 비교해보고 비슷한놈이라고 판단하는 간단한 로직밖에는 없으니까요.
> result <- knn(train, test, wbcd[1:500, c("diagnosis")], k=3)
> result
[1] Malignant Malignant Malignant Benign Benign Benign Benign Benign
[9] Malignant Malignant Benign Malignant Malignant Benign Malignant Benign
[17] Malignant Malignant Malignant Benign Benign Benign Malignant Benign
[25] Benign Benign Benign Malignant Benign Benign Benign Benign
[33] Benign Malignant Malignant Benign Benign Benign Benign Benign
[41] Malignant Benign Benign Malignant Malignant Benign Benign Benign
[49] Benign Benign Benign Benign Malignant Benign Benign Malignant
[57] Benign Benign Benign Benign Benign Benign Benign Benign
[65] Benign Benign Malignant Benign Malignant
Levels: Benign Malignant
예측은 과연 잘한것인가? 예측모델 평가하기
뭔가를 열심히 예측하기는 했는데, 이로써는 제대로 예측한건지 알수가 없습니다. 기존에 있던 데이터와 한번 대조해볼수 있습니다.
test_label <- wbcd[501:569,c("diagnosis")]
table(test_label, result)
result
test_label Benign Malignant
Benign 45 0
Malignant 2 22
참부정 거짓긍정 ?? 도대체 무슨말이야?
위의 테이블은 좀더 있어보이는 말로 혼동행렬이라고 부르는데, 실제 암덩어리로 구별된 놈들이 진짜 암덩어리인지 쉽게 볼수있는 간단한 테이블입니다. 이런 혼동행렬에는 True Negative, True Positive, False Positive, False Negative 등의 명칭이 있습니다.
아무리 다시봐도 헷갈리는데, 이를 한글로 번역해도 좀 이상합니다. 참긍정, 참부정, 거짓긍정, 거짓부정. 여기서 참긍정은 맞는것을 맞다고 하는것이고 참부정은 틀린걸 틀리다고 하는것입니다.
결국 '참'은 항상 예측결과가 맞다는 뜻이며, 거짓긍정의 경우에는 맞다고 우겼지만 사실은 아니고, 거짓부정의 경우에는 아니라고 우겼지만 사실은 맞는 경우입니다. 한번 곱씹어보세요. 참은 항상 진실만을 이야기한다는것을 잊지 마세요.
여기서 긍정이란, 문맥에 따라 달라지며 암덩이가 있다고 판별한결과를 긍정이라고 둔 경우, 참긍정은 실제로 있는 경우고 거짓긍정은 없는데 있다고 한 경우입니다.
그렇지만 제일 문제가 되는것은 거짓부정, 즉 없다고 했는데 실제로는 있는 경우이며 이러한경우에는 암을 진단못한채로 영문도 모른채 죽어갈수도 있어서, 참긍정보다 (정밀검진의 대상) 훨씬 위험이 크다고 할수 있겠습니다.
혼동행렬 자세히 보기
gmodels패키지 안에 들어있는 CrossTable() 함수는 혼동행렬을 조금 더 자세하게 표현해줍니다.
library(gmodels)
CrossTable(test_label, result)
| result
test_label | Benign | Malignant | Row Total |
-------------|-----------|-----------|-----------|
Benign | 45 | 0 | 45 |
| 6.716 | 14.348 | |
| 1.000 | 0.000 | 0.652 |
| 0.957 | 0.000 | |
| 0.652 | 0.000 | |
-------------|-----------|-----------|-----------|
Malignant | 2 | 22 | 24 |
| 12.593 | 26.902 | |
| 0.083 | 0.917 | 0.348 |
| 0.043 | 1.000 | |
| 0.029 | 0.319 | |
-------------|-----------|-----------|-----------|
Column Total | 47 | 22 | 69 |
| 0.681 | 0.319 | |
-------------|-----------|-----------|-----------|
실제로, 실제로 암이 진단된 경우에 정말 암이 있을 확률 (100%), 정상이라고 판단했는데도 암이 있을 확률 (4.3%) 등등을 자세히 살펴볼 수 있어 종종 쓰입니다.
몇가지 더 알아보자 : 데이터 수치 조절하기
데이터를 0~1로 조절해 정규화하기
실제로 어떤 데이터 항목의 분포가 0~0.000001이고, 어떤 항목은 0~99999999 라면 전자의 항목의 영향력은 미미합니다. 실제로 그 항목이 중요한데도 말이죠. 예를들어 조금의 방사능이라도 판별해내는것이 중요하다면 이렇게 수치가 극단적으로 작은것들을 정규화 해줄 필요가 있습니다. 하나의 간단한 함수를 만들수 있는데,
normalize <- function(x) {
return ((x - min(x)) / (max(x) - min(x)))
}
위의 함수로 apply()를 써서 적용시키면, 모든 수치가 조절이 됩니다.
데이터를 Z 표준점수로 정규화하기
normalize대신, 실제로 각각의 독립변수들이 정규분포를 따르고 있다면 이를 Z표준점수로 표준화 시켜줄수 있습니다. 이를 가능케해주는 함수명은 scale() 함수입니다.
scale(wbcd)
시리즈로 글을 써나갈 예정이라면 #kr-series 태그도 써보심이 어떨까요? 전 예전 #kr-python 이라는 태그도 몇 번 썼으니, #kr-r 이라는 태그를 주도하시는 것도 어떨까 합니다 :)
그리고 전 편의 링크를 후편에 적어놓으면 필요한 사람은 또 찾아갈 수 있는 효과가 있습니다.
시리즈물로 올리려 합니다~ 모든글은 직접 적었고, 다른 소스는 없습니다 ㅎㅎ 보시는것 같으면 계속해서 올릴게요
오 재밌습니다. ㅎㅎㅎ ^^ 하하 아 이거 써먹기 좋을 것 같아요. 고맙습니다.
감사합니다~ 스티밋에 오랜만에 오니 적응이 잘 안되네요 ㅋㅋ
잘 하시는 거 같은데요. ㅎㅎㅎㅎ 내용 좋네요. 열심히 배우겠습니다.