Atomic And Molecular Clusters Latest Preprints | 2019-05-02
Atomic And Molecular Clusters
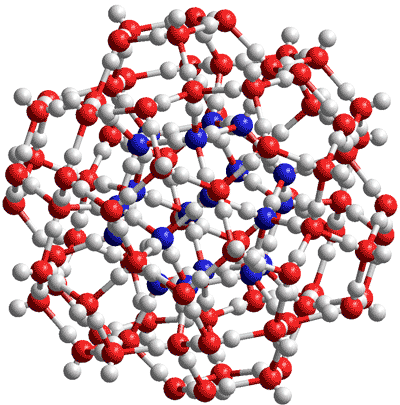
Reliability of two Embedded Atom Models for the Description of Ag@Au Nanoalloys (1904.10817v2)
Marta Bon, Nabeel Ahmad, Rolf Erni, Daniele Passerone
2019-04-24
The validation of embedded atom models (EAM) for modelling nanoalloys requires to verify both a faithful description of the individual phases and a convincing scheme for the mixed interactions. In this work, we present a systematic benchmarking of two widely adopted EAM parameterizations, i.e. by Foiles [S. M. Foiles et al. Phys. Rev. B 33, 7983 (1986)] and by Zhou [X. W. Zhou et al. Phys. Rev. B, 69, 144113 (2004)] with density functional theory calculations for the description of processes at Ag@Au nanoalloys surfaces and nanoclusters.
An Improved Descriptor of Cluster Stability. Application to Small Carbon Clusters (1904.11317v1)
Jose I. Martinez, Julio A. Alonso
2019-04-25
The mass spectra of gas-phase clusters in cluster beams have a rich structure where the relative heights of the peaks compared to peaks corresponding to clusters of neighbor sizes reveal the stability of the clusters as a function of the size
. In an analysis of the published mass spectrum of carbon clusters cations
with
16 we have employed the most common descriptor of cluster stability, which is based on comparing the total energy of the cluster of size
Efimov universality with Coulomb interaction (1901.03643v2)
C. H. Schmickler, H. -W. Hammer, E. Hiyama
2019-01-11
The universal properties of charged particles are modified by the presence of a long-range Coulomb interaction. We investigate the modification of Efimov universality as a function of the Coulomb strength using the Gaussian expansion method. The resonant short-range interaction is described by Gaussian potentials to which a Coulomb potential is added. We calculate binding energies and root mean square radii for the three- and four-body systems of charged particles and present our results in a generalised Efimov plot. We find that universal features can still be discerned for weak Coulomb interaction, but break down for strong Coulomb interaction. The root-mean-square radius plateaus at increasingly smaller values for strong Coulomb interaction and the probablity distributions of the states become more concentrated inside the Coulomb barrier. As an example, we apply our universal model to nuclei with an alpha-cluster substructure. Our results point to strong non-universal contributions in that sector.
Pure Molecular Beam of Water Dimer (1904.08716v2)
Helen Bieker, Jolijn Onvlee, Melby Johny, Lanhai He, Thomas Kierspel, Sebastian Trippel, Daniel A. Horke, Jochen Küpper
2019-04-18
Spatial separation of water dimer from water monomer and larger water-clusters through the electric deflector is presented. A beam of water dimer with
purity and a rotational temperature of
K was obtained. Following strong-field ionization using a
fs laser pulse with a wavelength centered around
nm and a peak intensity of
we observed proton transfer and
of the ionized water dimer broke apart into a hydronium ion
and OH.
Time-resolved Spectroscopy of Interparticle Coulombic Decay Processes (1904.05861v2)
Elke Fasshauer, Lars Bojer Madsen
2019-04-11
We report theory for time-resolved spectator resonant Interparticle Coulombic Decay (ICD) processes. Following excitation by a short XUV pulse, the spectrum of the resonant ICD electron develops. Strong-field ionization quenches the decay at different time delays and initiates regular ICD, whose ICD electron signal can be measured without interference effects. The typical lifetimes of ICD processes allow for the observation of oscillations of the time- and energy-differential ionization probability. We propose to utilize this oscillation to measure lifetimes of electronic decay processes.
Prediction of activation energy barrier of island diffusion processes using data-driven approaches (1902.10282v2)
Shree Ram Acharya, Talat S. Rahman
2019-02-22
We present models for prediction of activation energy barrier of diffusion process of adatom (1-4) islands obtained by using data-driven techniques. A set of easily accessible features, geometric and energetic, that are extracted by analyzing the variation of the energy barriers of a large number of processes on homo-epitaxial metallic systems of Cu, Ni, Pd, and Ag are used along with the activation energy barriers to train and test linear and non-linear statistical models. A multivariate linear regression model trained with energy barriers for Cu, Pd, and Ag systems explains 92% of the variation of energy barriers of the Ni system, whereas the non-linear model using artificial neural network slightly enhances the success to 93%. Next mode of calculation that uses barriers of all four systems in training, predicts barriers of randomly picked processes of those systems with significantly high correlation coefficient: 94.4% in linear regression model and 97.7% in artificial neural network model. Calculated kinetics parameters such as the type of frequently executed processes and effective energy barrier for Ni dimer and trimer diffusion on the Ni(111) surface obtained from KMC simulation using the predicted (data-enabled) energy barriers are in close agreement with those obtained by using energy barriers calculated from interatomic interaction potential.
Quantum entanglement between two magnon modes via Kerr nonlinearity (1904.04167v1)
Zhedong Zhang, Marlan O. Scully, Girish S. Agarwal
2019-04-08
We propose a scheme to entangle two magnon modes via Kerr nonlinear effect when driving the systems far-from-equilibrium. We consider two macroscopic yttrium iron garnets (YIGs) interacting with a single-mode microcavity through the magnetic dipole coupling. The Kittel mode describing the collective excitations of large number of spins are excited through driving cavity with a strong microwave field. We demonstrate how the Kerr nonlineraity creates the entangled quantum states between the two macroscopic ferromagnetic samples, when the microcavity is strongly driven by a blue-detuned microwave field. Such quantum entanglement survives at the steady state. Our work offers new insights and guidance to designate the experiments for observing the entanglement in massive ferromagnetic materials. It can also find broad applications in macroscopic quantum effects and magnetic spintronics.
Few-body quantum method in a 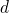 -dimensional space (1901.02667v2)
-dimensional space (1901.02667v2)
E. Garrido, A. S. Jensen, R. Álvarez-Rodríguez
2019-01-09
In this work we investigate the continuous confinement of quantum systems from three to two dimensions. Two different methods will be used and related. In the first one the confinement is achieved by putting the system under the effect of an external field. This method is conceptually simple, although, due to the presence of the external field, its numerical implementation can become rather cumbersome, especially when the system is highly confined. In the second method the external field is not used, and it simply considers the spatial dimension
A tight-binding model for the excitonic band structure of a one-dimensional molecular chain: UV-Vis spectra, Zak phase and topological properties (1904.01881v1)
Wei Wu, Jin Zhang
2019-04-03
Recently organic optics becomes a hot topic due to the rapid development of organic light-emitting diodes, organic solar cells, and organic photon detectors. The optical spectra of the molecular semiconductors are difficult to solve an model from first-principles because (i) the very large number of atoms in a unit cell and (ii) the accurate theoretical excited state is still under development. Here we present a tight-binding model of an exciton band structure in a molecular chain. We take into account the intra-molecule and charge-transfer excitation within a molecular dimer in a unit cell, then we apply the tight-binding model by including the coupling between two types of excitations. We not only found that our calculations can explain a body of UV-Vis optical spectra of transition-metal phthalocyanines, but also a one-dimensional excitonic topological band structure if we fine-tune the couplings in a dimerized molecular chain. We have found a large space to obtain the topological Zak phase in the parameter space, in which there is a simple linear relationship between the hopping integrals between cells and within cell.
Stretched or noded orbital densities and self-interaction correction in density functional theory (1903.00611v2)
Chandra Shahi, Puskar Bhattarai, Kamal Wagle, Biswajit Santra, Sebastian Schwalbe, Torsten Hahn, Jens Kortus, Koblar A. Jackson, Juan E. Peralta, Kai Trepte, Susi Lehtola, Niraj K. Nepal, Hemanadhan Myneni, Bimal Neupane, Santosh Adhikari, Adrienn Ruzsinszky, Yoh Yamamoto, Tunna Baruah, Rajendra R. Zope, John P. Perdew
2019-03-02
Semi-local approximations to the density functional for the exchange-correlation energy of a many-electron system necessarily fail for lobed one-electron densities, including not only the familiar stretched densities but also the less familiar but closely-related noded ones. The Perdew-Zunger (PZ) self-interaction correction (SIC) to a semi-local approximation makes that approximation exact for all one-electron ground- or excited-state densities and accurate for stretched bonds. When the minimization of the PZ total energy is made over real localized orbitals, the orbital densities can be noded, leading to energy errors in many-electron systems. Minimization over complex localized orbitals yields nodeless orbital densities, which reduce but typically do not eliminate the SIC errors of atomization energies. Other errors of PZ SIC remain, attributable to the loss of the exact constraints and appropriate norms that the semi-local approximations satisfy, and suggesting the need for a generalized SIC. These conclusions are supported by calculations for one-electron densities, and for many-electron molecules. While PZ SIC raises and improves the energy barriers of standard generalized gradient approximations (GGA's) and meta-GGA's, it reduces and often worsens the atomization energies of molecules. Thus PZ SIC raises the energy more as the nodality of the valence localized orbitals increases from atoms to molecules to transition states. PZ SIC is applied here in particular to the SCAN meta-GGA, for which the correlation part is already self-interaction-free. That property makes SCAN a natural first candidate for a generalized SIC.