Alteraciones congénitas y adquiridas de la coagulación: revisión del tema.
Hola Steemians, En esta ocasión estudiaremos el tema de las coagulopatías; tantos las congénitas como las adquiridas. para entender mejor cada entidad clínica es necesario conocer la fisiología de la hemostasia y la coagulación.
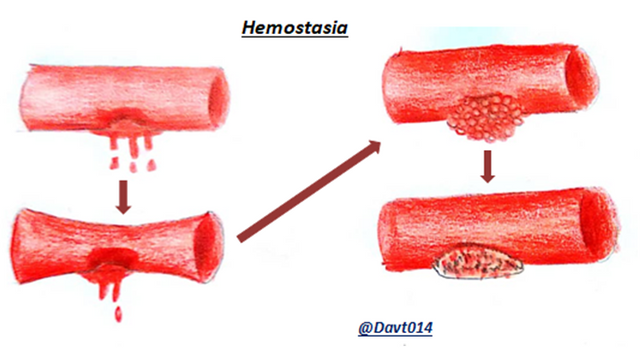

Hemostasia y coagulación
La hemostasia depende básicamente de 3 factores:
Endotelio: Su función es comandar la relación vasodilatación-vasoconstricción hacia el balance, regula la síntesis y traslado de los miocitos (células musculares) de la pared de los vasos sanguíneos y también regula la hemostasia.
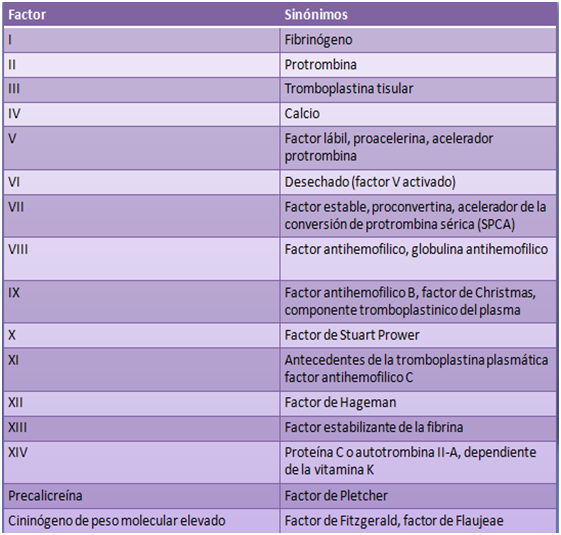
Factores de coagulación: son aquellas sustancias plasmáticas; en su mayoría de naturaleza proteica, cuya función como su nombre lo indica es la activación o regulación de la coagulación sanguínea.

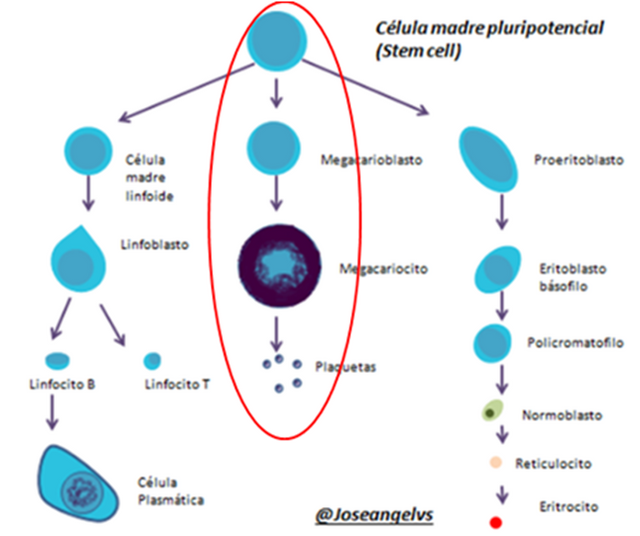
- Plaquetas: no son células, sino más bien fracciones celulares. Tienen diferentes funciones, pero en este tema solo nos importa saber que son las responsables de formar el “tapón” plaquetario, una de las primeras etapas de la coagulación.
Las plaquetas son sintetizadas en la médula ósea a partir de la célula madre pluripotencial, son el resultado final de la línea celular megacariocitica
Etapas de la hemostasia
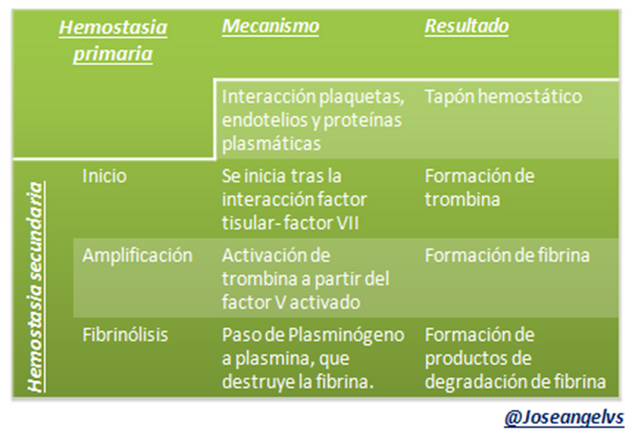
La hemostasia ocurre en dos etapas con objetivos diferentes: hemostasia primaria y secundaria.
Hemostasia primaria:
Como se mencionó anteriormente el endotelio es un factor regulador muy importante de la hemostasia. En condiciones normales libera sustancias “anticoagulantes” por así decirlo, y vasodilatadoras como serían el Óxido nítrico, Heparán y eicosanoides con función antiagregante plaquetario como la prostaciclina.
Al existir alguna noxa que afecte la integridad del vaso sanguíneo se liberan sustancias pro-coagulantes como la endotelina y la angiotensina que producen vasoconstricción para evitar mayor salida de sangre activando así la Hemostasia primaria.
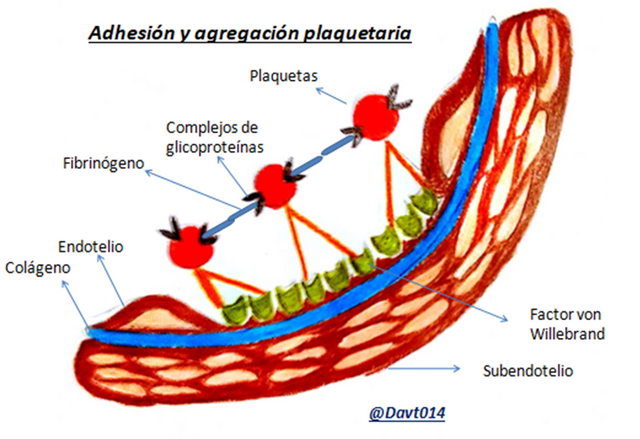
Lo primero que ocurre es la adhesión plaquetaria; las plaquetas a través de unas proteínas presentes en su membrana (glicoproteína lb) se unen al colágeno que está en los vasos sanguíneos, todo esto con la ayuda del factor von Willebrand.
Luego de la adhesión, las plaquetas se “activan” dando las siguientes consecuencias:
- Liberan sustancias (serotonina, adenosín difosfato, calcio, etc) que atraen y activan otras plaquetas, dichas sustancias se encuentran en los gránulos dentro de las plaquetas.
Elaboración y liberación de eicosanoides vasoconstrictores como el Tromboxano A2.
En la membrana de las plaquetas se encuentran algunos fosfolípidos con cargas negativas, esto hace que el factor X se adhiera a las plaquetas para activarse en la cascada de coagulación.
La glicoproteína IIb cambia de forma volviéndose activa (glicoproteína IIIa).
Luego de la activación plaquetaria se promueve la ”agregación plaquetaria” formándose puentes entre las plaquetas mediante las glicoproteínas y fibrinógeno.
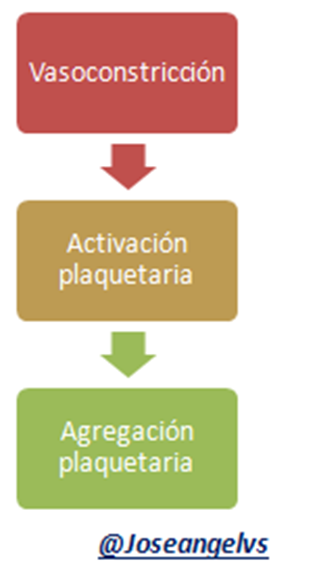
Hemostasia secundaria
Esta etapa tiene como objetivo la creación de un coágulo de fibrina (Ia) estable. Para esto se requiere la activación de los factores de coagulación de forma secuencial hasta lograr el factor II activado (trombina, IIa), cuya función es degradar al fibrinógeno a fibrina. Además para lograr la coagulación se requiere la intervención de otras sustancias como el calcio iónico y Fosfolipidos.
El factor II (trombina) activa a otros factores como el I, V, VIII, XIII y sustancias limitantes como sería la proteína C.
Para que exista un control se requieren sustancias que Inhiban la coagulación como la antitrombina que inhiben al factor X y a la trombina, proteína S y C que inhiben al factor V y VIII.
Cascada de coagulación
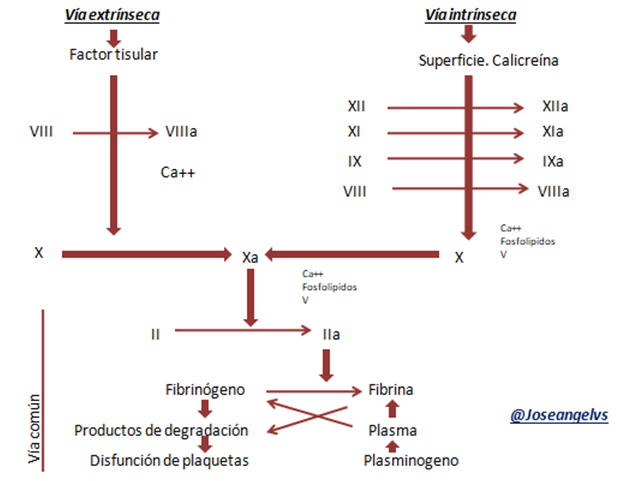
Está dada por 3 vías: la intrínseca, extrínseca y la vía común.
- Vía intrínseca: también llamada “vía por contacto” ya que esta se inicia cuando la sangre toca alguna superficie extraña diferente a la pared interna de los vasos sanguíneos (endotelio); las fibras de colágeno presentes cuando hay alguna ruptura del vaso sanguíneo es suficiente para desencadenar este proceso. Se llama intrínseca porque requiere factores que están presentes en la sangre y anteriormente se pensaba que con solo esta vía era suficiente para desencadenar y culminar la coagulación, sin embargo hoy día se sabe que esta vía es más bien sirve para amplificar el proceso.
Se inicia con la activación de XII – XI- IX – VIII de forma secuencial, luego se forma el complejo "tenasa intrínseca" que interacción con los Fosfolipidos activando así el factor X.
Vía extrínseca: Se llama de esta forma ya que para poder iniciarse requiere factores que no se encuentran en la sangre. La sangre al tocar tejidos afectados se une a un complejo de factores como el factor tisular (también se llama vía del factor tisular) formando la "tenasa extrínseca" activando rápidamente al factor X.
Vía común: las dos vías, tanto la intrínseca como la extrínseca convergen en la vía común, cuyo fin es la es la transformación de fibrinógeno en fibrina y la estabilización del coágulo.
Gracias a ambas tenasas se activa el factor X el cual transforma a la protrombina en trombina, que es el que transforma el fibrinógeno en fibrina.
Fibrinólisis
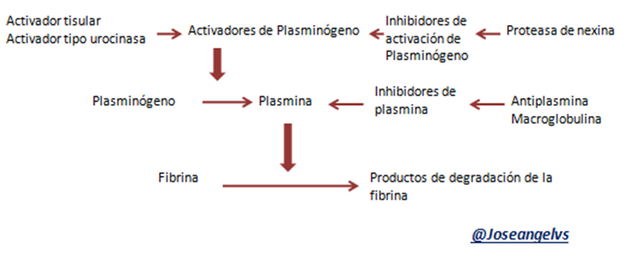
Para evitar la formación de trombos debe existir una forma de ir destruyendo esos coágulos que se van formando. El t-PA (activador de plasminógeno tisular) y factores como XII y el sistema calicreina-quinina hacen que se active la plasmina, cuya función es destruir la fibrina formada. La fibrinólisis se va activando al mismo tiempo que se están formando los coágulos.
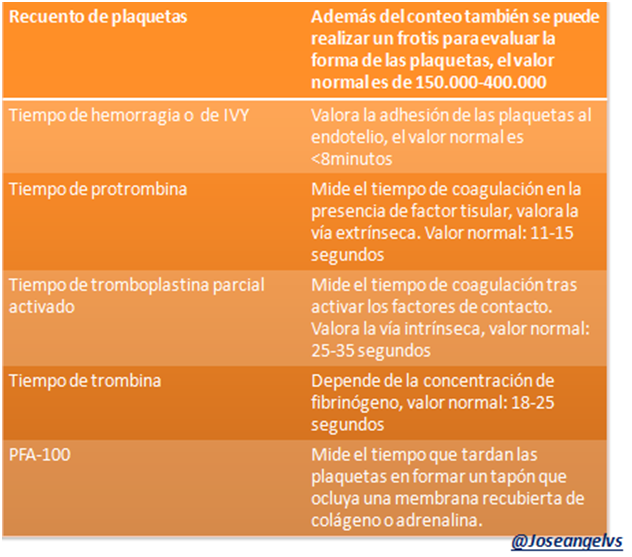
Pruebas básicas para la valoración la hemostasia
En la práctica clínica es importante conocer las diferentes pruebas para valorar la coagulación para poder establecer los posibles diagnósticos.
La hemostasia puede ser valorada por “pruebas globales” que nos orientarían a que el sistema en general está alterado pero no cual componente del sistema lo está. Y las “pruebas específicas” que miden de forma independiente cada componente, por ejemplo cuantificar algún factor de coagulación.
De forma didáctica también podemos dividir el estudio de la siguiente manera:
Hemostasia primaria: Contaje de plaquetas, tiempo de hemorragia, se puede inducir la agregación y adhesión plaquetaria, determinación de factores como el de von Willebrand.
Hemostasia secundaria: se pueden usar los tiempos de trombina y protrombina, determinación del fibrinógeno, cuantificación de los factores de coagulación.
Trastornos congénitos de la coagulación
Son un grupo variado de enfermedades que se caracterizan por alteración en la coagulación, esto se debe a variaciones en la cantidad o calidad de los factores de la coagulación, consecuencia a alteraciones ocurridas durante el embarazo o defecto genético hereditario.
Enfermedad de von Willebrand
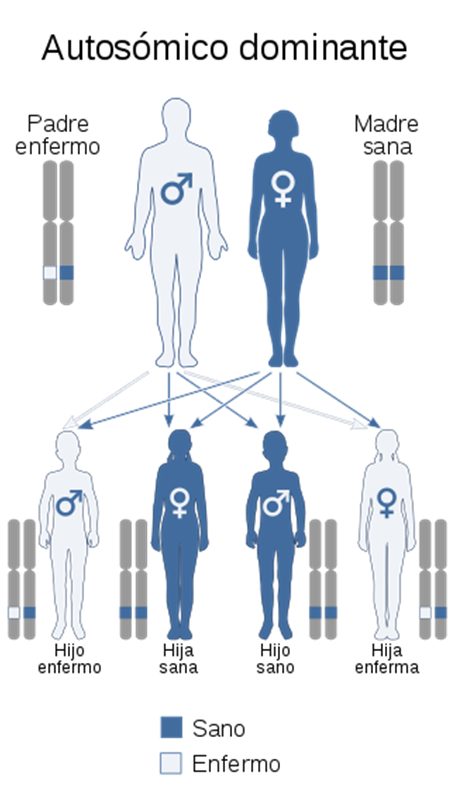
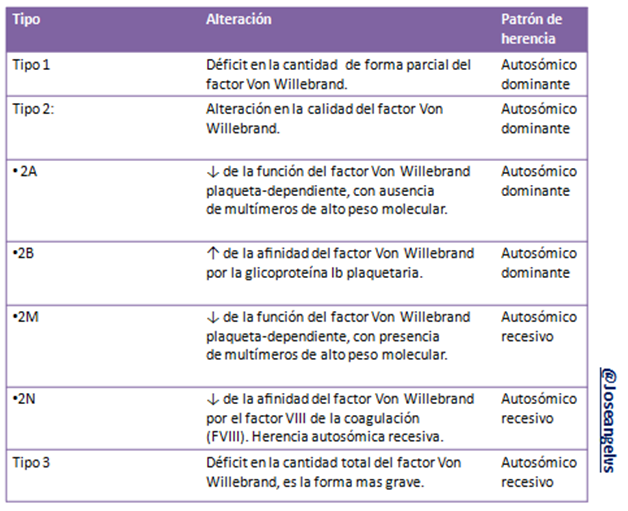
Es una enfermedad congénita aunque puede ser adquirida como consecuencia de otra anomalía (lupus eritematoso sistémico o hipernefroma). Se debe a alteraciones en la cantidad o calidad del factor von Willebrand, de estas coagulopatías congénitas es la más común y generalmente tiene un patrón de herencia autosómico dominante.
{kind=link}
Clasificación
Manifestaciones clínicas
La severidad puede variar dependiendo del tipo, sin embargo las formas leves son las más comunes, lo característico es la aparición de hemorragias posterior a lesiones cortantes o procedimientos invasivos (como extracción de una pieza dental). El sangrado normalmente es cutaneomucoso aunque puede ser también muscular o dentro de las articulaciones.
Diagnóstico
- El factor von Willebrand se puede determinar en sangre, cuyos niveles se encuentran alterados.
- El tiempo de hemorragia se encuentra aumentado.
- Tiempo parcial de tromboplastina activado se encuentra aumentado, asociado a un tiempo de protrombina normal.
- El estudio de la agregación plaquetaria se encuentra alterado, con una disminución de la misma.
- Tomar en cuenta la historia familiar para determinar algún patrón de herencia.
Tratamiento
- No requiere un tratamiento continuo, en este sentido solo se tratan los episodios hemorrágicos o profilaxis preventiva antes de alguna cirugía.
- En primer lugar se debe hacer medidas locales de compresión.
- Farmacoterapia: se debe usar en orden, lo primero son los fármacos locales como la goma de fibrina, en caso de que no sea suficiente se puede usar ácido tranexámico que es un antifibrinolítico, si sigue la hemorragia se usa desmopresina que solo es útil en la tipo 1 ya que aumenta la liberación del factor von Willebrand.
- También están indicados los concentrados del factor von Willebrand.
Hemofilias
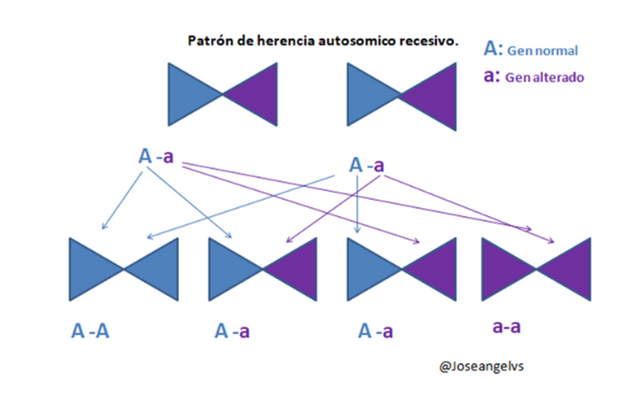
Es un trastorno genético que afecta la coagulación, su patrón de herencia es ligado al cromosoma X aunque puede ser autosómico recesivo (el tipo C).
Clasificación
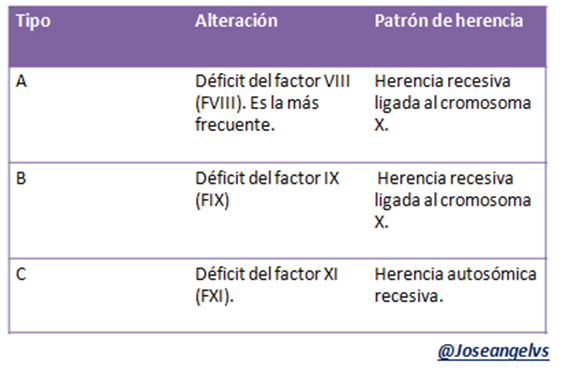
Se puede clasificar dependiendo de la gravedad del déficit:
- Hemofilia leve: >5% de déficit del factor asociado, es la forma más común, se manifiesta como hemorragias en cirugías mayores o traumatismo graves.
- Hemofilia moderada: el déficit del factor es de 1-5%, se manifiesta como hemorragias en cirugías o traumatismo pequeños.
- Hemofilia severa: el déficit <1%, las hemorragias son espontaneas o se deben a traumatismo mínimos.
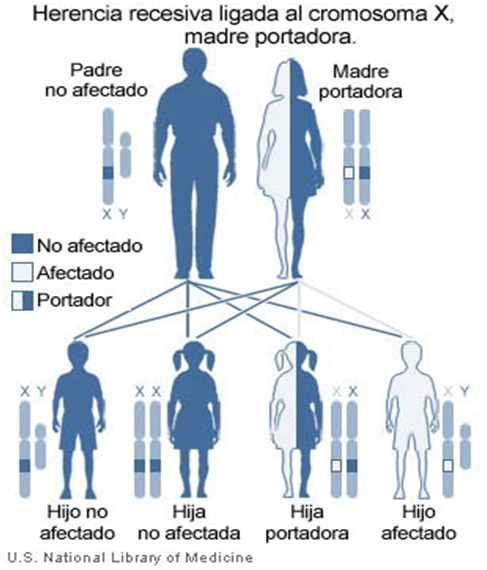
Los hombres pueden ser enfermos o sanos, pero en las mujeres existe la figura de “portadora” que no presentan sintomatología, sin embargo tienen menos cantidad del factor von Willebrand.
Manifestaciones clínicas
- En el 90% de los casos el síntoma predominante es el sangrado en las articulaciones especialmente en la rodilla, aunque puede aparecer en codos, hombros o tobillos.
- Sangrado intramuscular.
- No hay petequias.
- Otros sangrados: dentro del cráneo, en el aparato respiratorio superior, hemorragias digestivas, en el sistema genitourinario.
Diagnóstico
Pruebas de laboratorio:
- Tiempo parcial de tromboplastina activado se encuentra aumentado que debería normalizarse al mezclar la sangre del paciente con otra sangre de una persona sana ya que contiene el factor que se está deficiente.
- Tiempo de trombina normal.
- Factor de coagulación se encuentra disminuido pero depende del tipo.
Estudios genéticos
- Para determinar el tipo específico y diagnósticos intrauterinos.
Tratamiento
- El tratamiento debe ser sintomático ya que no existe una cura definitiva; aunque se han tenido resultados positivos con de trasplante de hígado.
- Farmacoterapia: antihemorrágicos locales, antifibrinolíticos.
- Concentrado sanguíneo que contenga el factor deficiente según el tipo.
- Está contraindicado el uso de ácido acetilsalicílico por sus acciones antiagregantes plaquetarios que pueden predisponer a hemorragias.
Trastornos adquiridos de la coagulación
Son un grupo variado de enfermedades que se caracterizan por alteración en la coagulación, esto se debe a variaciones en la cantidad o calidad de los factores de la coagulación, como consecuencia a afecciones secundarias o inmunitarias que afecten al sistema hemostático.
Coagulación intravascular diseminada
Es un síndrome clínico caracterizado por múltiples hemorragias, debido a un consumo de los factores de coagulación secundaria a la producción exagerada de trombina, que ocasiona una formación de excesiva de coágulos además de la hemorragia.
Lo más importante de este síndrome es diagnosticar y tratar la causa, que pueden ser:
- Infecciones severas, sobre todo por bacilos gramnegativos.
- Traumatismos graves, sobre todo craneoencefálicos ya que hay elevación de los fosfolípidos.
- Tumores malignos, como la leucemia promielocítica, ya que los gránulos de estas células malignas producen sustancias que inducen coagulación.
- Anemia hemolítica microangiopatica.
- Traumas obstétricos: enfermedad hipertensiva del embarazo, placenta previa, desprendimiento prematuro de placenta, etc. En estos casos puede haber aumento del factor tisular promoviendo la coagulación.
- Venenos de animales: algunas serpientes producen venenos que afectan la coagulación.
Manifestaciones clínicas
- Múltiples hemorragias.
- Trombosis de grandes vasos.
- Fiebre.
- Shock secundario a afectación de la microvasculatura.
Diagnóstico
El diagnóstico es clínico, ya que no hay pruebas que aseguren el diagnostico sin embargo hay que tener ciertas consideraciones:
- Disminución de las plaquetas, aumento del tiempo de trombina y el de tromboplastina parcialmente activado.
- Aumento del “dímero D”, disminución del fibrinógeno, y aumento de los productos de desecho consecuencia de la fibrinólisis.
Tratamiento
Lo principal es el tratamiento de la causa desencadénate, sin embargo es importante el uso de transfusiones sanguinas tipo plasma fresco congelado o crioprecipitado para reponer los factores perdidos.
Se puede usar tratamiento anticoagulantes a dosis bajas tipo heparina para evitar las trombosis.
Síndrome antifosfolípido
Es un síndrome auto-inmunitario caracterizado por trombosis tanto venosas como arteriales de forma recurrente, abortos recurrentes, disminución de las plaquetas o anemia hemolítica y la presencia de auto-anticuerpos antifosfolípidos.
Los auto-anticuerpos antifosfolípidos inhiben la función de estos, por lo que las plaquetas promueven la coagulación produciendo síndromes tromboticos mas no hemorrágicos.
Los auto-anticuerpos antifosfolípidos más conocidos son:
- Anticardiolipina.
- Beta glicoproteína
- Anticoagulante lúpico
Tratamiento
Consiste en evitar los eventos tromboticos administrando agentes antiagregantes plaquetarios como ácido acetilsalicílico o anticoagulantes como warfarina o heparina.
Referencias
- Fauci AS, et al. Harrison’s principles of internal medicine. Vol 2. 19th ed. New York: McGraw Hill; 2015
- Lichtman A, et al. Williams Manual de hematologia. 8va ed: McGraw Hill; 2014.

Muy bien explicado. Una síntesis excepcional!
Gracias por leer doc.
Congratulations! This post has been upvoted from the communal account, @minnowsupport, by Joseangelvs from the Minnow Support Project. It's a witness project run by aggroed, ausbitbank, teamsteem, theprophet0, someguy123, neoxian, followbtcnews, and netuoso. The goal is to help Steemit grow by supporting Minnows. Please find us at the Peace, Abundance, and Liberty Network (PALnet) Discord Channel. It's a completely public and open space to all members of the Steemit community who voluntarily choose to be there.
If you would like to delegate to the Minnow Support Project you can do so by clicking on the following links: 50SP, 100SP, 250SP, 500SP, 1000SP, 5000SP.
Be sure to leave at least 50SP undelegated on your account.
Interesante abordaje al proceso de coagulación @joseangelvs.
Gracias por leer, el tema de la coagulación es algo difícil por la cantidad de factores que implica
Excelente @joseangelvs, realmente no es fácil abarcar todos los detalles.
Gracias por leer doc, si un tema algo difícil de desarrollar.
Amigo el tema de coagulación es un tema muy extenso pero en el post lo explicaste super detallado y claro, te felicito
Gracias, que bueno que te gustara :)
Bastante puntual tu publicación. Una pregunta: ¿esta forma de organizar contenidos de medicina tiene un nombre? Lo pregunto porque tengo entendido que los médicos y profesionales afines tienen que ser concretos a la hora de hablar y ello se nota en esta publicación a través de las listas y series de subtítulos, y en las publicaciones de otros steemians que también crean contenido para esta área.
Buen post!
Hola gracias por leer, bueno realmente no se si tenga un nombre esta forma de organizar la publicación, mi estilo es siempre hablar de lo fisiológico de manera que la o las patologías que mencionaré sean mejor entendidas.
Entiendo... Él estilo de los post de ustedes los médicos es muy práctico!
Excelente post Jose, como siempre preciso y conciso algo clave para entender muchas patologías. Gracias por compartirlo.
Gracias Miguel muy pendiente como siempre.
Muy interesante el tema y qué buena manera de sintetizar, gracias por compartir
gracias por leer, saludos.
Muy bien elaborado y el contenido se entiende a la perfección
gracias doc, era lo que mas temía que no se entendiera :)
sumamente interesante uno de los temas que siempre he tenido respeto es a este a las cuagulopatias es un tema que lo no importa cuanto lo leas siempre sera como el primer dia de lo leas
Gracias por leer, tienes razón este tema hay que estar repasándolo siempre.